
Huntington’s, Alzheimer's and Parkinson’s - New Hope?
Randal Halfmann, a renowned researcher in the field of molecular biology, has recently made a significant contribution to the study of amyloid — a protein that is implicated in various diseases, including Alzheimer's, Huntington's and Parkinson's. Halfmann's research, which was published on June 13, 2023, in the prestigious scientific journal eLife, sheds new light on the mechanisms underlying the formation of amyloid.
For the first time, with other scientists at the Stowers Institute for Medical Research, he has uncovered the structure of the first step in amyloid formation, called the nucleus. They discovered the nucleus forms within a single protein molecule. They have also identified 36 glutamine repeats (Qs — glutamine: abbreviated as Q) as the crucial number needed for nucleus formation in a protein sequence, offering a specific target for potential therapeutic treatments of Huntington’s.[1]
The results of the study provide fresh insights into how these protein clumps form, and could eventually lead to the development of new treatments for amyloid-related diseases.
Amyloid formation
Macroscopic protein deposits of amyloids are associated with diseases such as Alzheimer’s (tau and β-amyloid) and Parkinson’s (⍺-synuclein). The issue with the protein is that it is much too big and typically misfolded, and passed a certain threshold (in length) that protein can become problematic, even toxic.
Until now, it was believed that amyloid fibrils were the problem behind the formation of plaques in the brain. However, attention shifted to the cytotoxicity of amyloid fibril precursors, notably amyloid oligomers, which appear to be the primary cause of toxicity.[2]
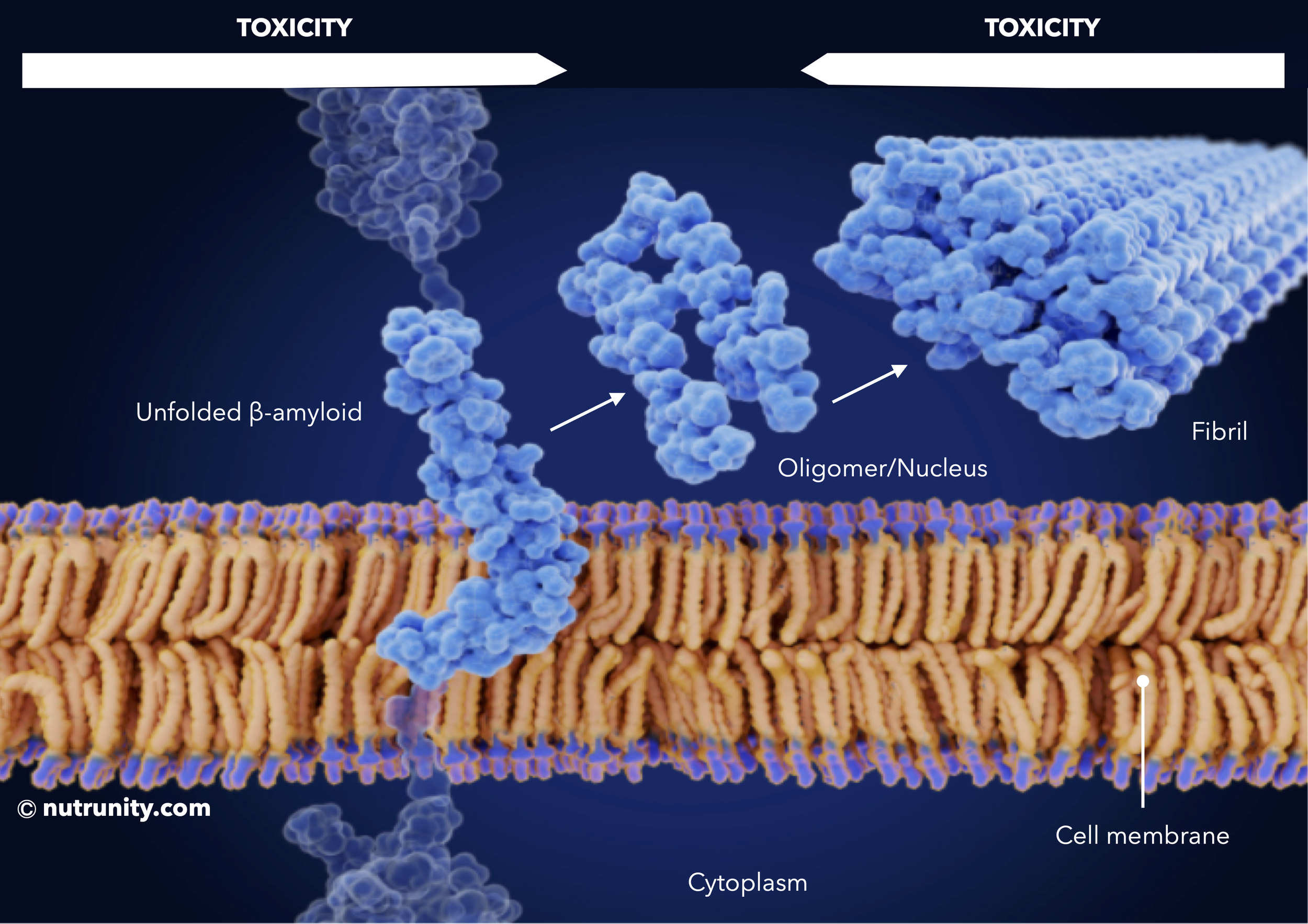
β-amyloid monomers self-assemble to form soluble toxic aggregates (oligomers), and finally into insoluble fibrils, which subsequently form cause synaptic dysfunction and neuronal death.
In biochemistry, an oligomer (from Greek oligo-: "a few" and -mer: "parts") is a molecular complex that consists of a few repeating units which could be derived from smaller molecules, or monomers (in this context, an unfolded peptide/protein: amyloid-β).
Oligomers then form fibrils and then amyloid plaque in a ‘cascade’ that leads to the development of Alzheimer’s disease. In this case, oligomers are also referred to as nuclei (singular: nucleus).
Polyglutamine (polyQ) refers to a protein segment that consists of multiple glutamine units. Polyglutamine-related illnesses are dominant genetic conditions that result in progressive neurodegeneration along with behavioural and physical impairments. This is particularly evident in Huntington's disease, often inherited (characterised by involuntary movements, progressive cognitive decline and emotional lability).[3]
In carriers, the polyQ tract can exceed 36 glutamines and may even go beyond 100 depending on the disease. These mutations are unstable and can lead to changes in repeat length between generations and within different cells and tissues of the same individual. Despite being expressed throughout the body ubiquitously, polyQ-containing proteins mainly cause pathological effects restricted to neuronal tissue.
Oligomers and neuronal activity
The aggregation of the β-amyloid peptide (a small chain of amino acids — smaller than a protein, a longer chain of amino acids) in some brain regions, particularly those involved in memory and cognition, is thought to be the decisive factor in the onset and progression of Alzheimer's disease.[4]
By crowding the synaptic cleft (also called synaptic gap, the space that separates two neurones, forming a junction between two or more neurons and helping nerve impulse pass from one neuron to the other), β-amyloid oligomers can interfere with neuronal activity and impulse. It is one of the pathways where memory and cognition can deeply be affected. Furthermore, this leads to the hyperactivation of the neurones and in the long term to neuronal and microglial inflammation.
Microglia, as the immune cells in the brain, play crucial roles in neuroinflammation and many cognitive disorders, including neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease.[5]
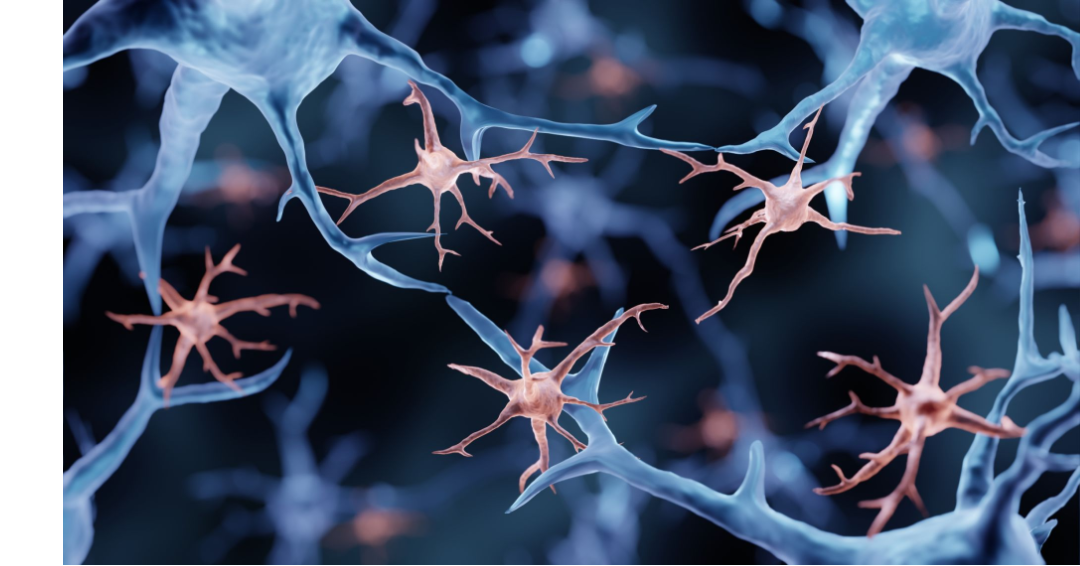
Microglia: brain resident immune cells that regulate brain development, maintenance of neuronal networks, and injury repair. They are involved in the uptake and clearance of amyloid-β (Aβ), as well as the formation of neuroinflammation
Note on microglia:
Microglia, the resident immune cells of the central nervous system, play a crucial role in maintaining brain function and responding to immune activity and inflammation. They constantly monitor their surroundings, detecting any signs of infection, injury, or abnormality. When activated, microglia swiftly respond, releasing various molecules and engulfing pathogens or damaged cells (phagocytosis).
Besides their role in immune defence, microglia also actively engage in synaptic remodelling, promoting neuronal connectivity and plasticity. This necessary activity ensures the maintenance of the neuronal network and healthy brain activity. Microglia-neuron junctions provide a communication channel whereby microglia can constantly monitor neuronal activation status and health to respond accordingly when disruptions occur. Disruption of this microglia-neuron communication channel synchronises and hyperactivates (hyperexcite) neurones, which is an underlying cause of seizures.[7-9].
However, dysregulated microglial activation can lead to chronic inflammation, potentially contributing to the development and progression of neurodegenerative disorders.
In Alzheimer's disease, microglia become activated and contribute to the neuroinflammatory response that is characteristic of the disease. While initially intended to protect, the excessive activation of microglia can actually exacerbate the pathology. Studies suggest that dysfunctional microglia fail to efficiently clear beta-amyloid plaques, the hallmark feature of Alzheimer's. Additionally, the chronic activation of microglia can lead to the release of toxic substances, further damaging surrounding neurones.
Microglia are also involved in the development of multiple sclerosis.[10] In the early stages, microglia may have a protective role by promoting tissue repair. However, long-term activation of microglia leads to neuroinflammation and the destruction of myelin, which insulates nerve fibres. Activated microglia release pro-inflammatory cytokines and free radicals, further exacerbating the immune response. Additionally, they participate in the recruitment and activation of other immune cells, such as T cells, which contribute to autoimmunity. However, recent research suggests that microglia
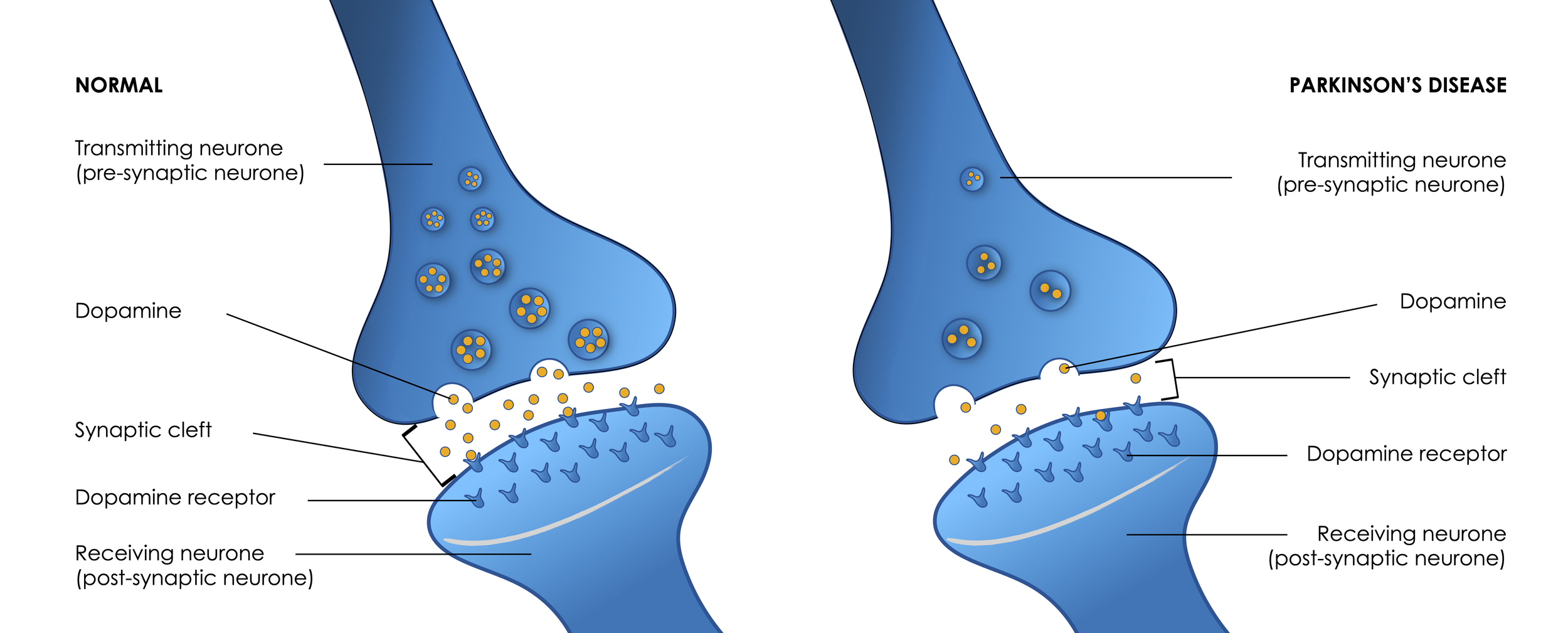
Synaptic cleft, dopamine dysfunction, a hallmark of Parkinson’s disease.
Looking at the illustration above, the synaptic cleft is free of unwanted components. Now imagine it being filled with amyloid oligomers, making it more difficult or preventing neurotransmitters from reaching the post-synaptic neurones
Another factor contributing to cognitive dysfunction includes the formation of plaques, which may restrict the exchange of nutrients and oxygen to the brain and the discharge of soluble β-amyloid peptides across the blood-brain barrier.
Astrocytes are a subtype of glial cells that make up the majority of cells in the human central nervous system, They perform metabolic, structural, homeostatic, and neuroprotective tasks such as clearing excess neurotransmitters, stabilizing and regulating the blood-brain barrier, and promoting synapse formation. Forming the blood-brain barrier, astrocytes only allow certain substances to cross the inner regions of the brain from the blood supply.

An astrocyte, forming an integral part of the blood-brain barrier

Astrocytes, the link between the blood system and neurones and synaptic performance.
Eventually, inflammation may lead to excessive oxidative stress (free radical production) and mitochondrial dysfunction and, ultimately, neuronal death and shrinkage of the cerebral cortex and hippocampus as observed in Alzheimer’s disease.[10]
Amyloid plaque and neurodegeneration
Imagine if you were revving the engine of your car by pressing the accelerator, but your hand brake was still fully engaged. You wouldn't be going anywhere, right? Instead, your tires could start to create black smoke and you may even cause increased wear and tear on your engine. If you’d continued pressing on the accelerator, the tires could eventually catch fire and you may even cause permanent damage to the engine. You may also run out of fuel.
This same concept applies to the brain. Overactivation of brain cells can lead to the release of toxic byproducts and damage to surrounding tissue. Neurones are sending impulses, but the information isn't relayed properly or may not reach the next neurones, leading to hyperactivity and eventually to neuronal death.
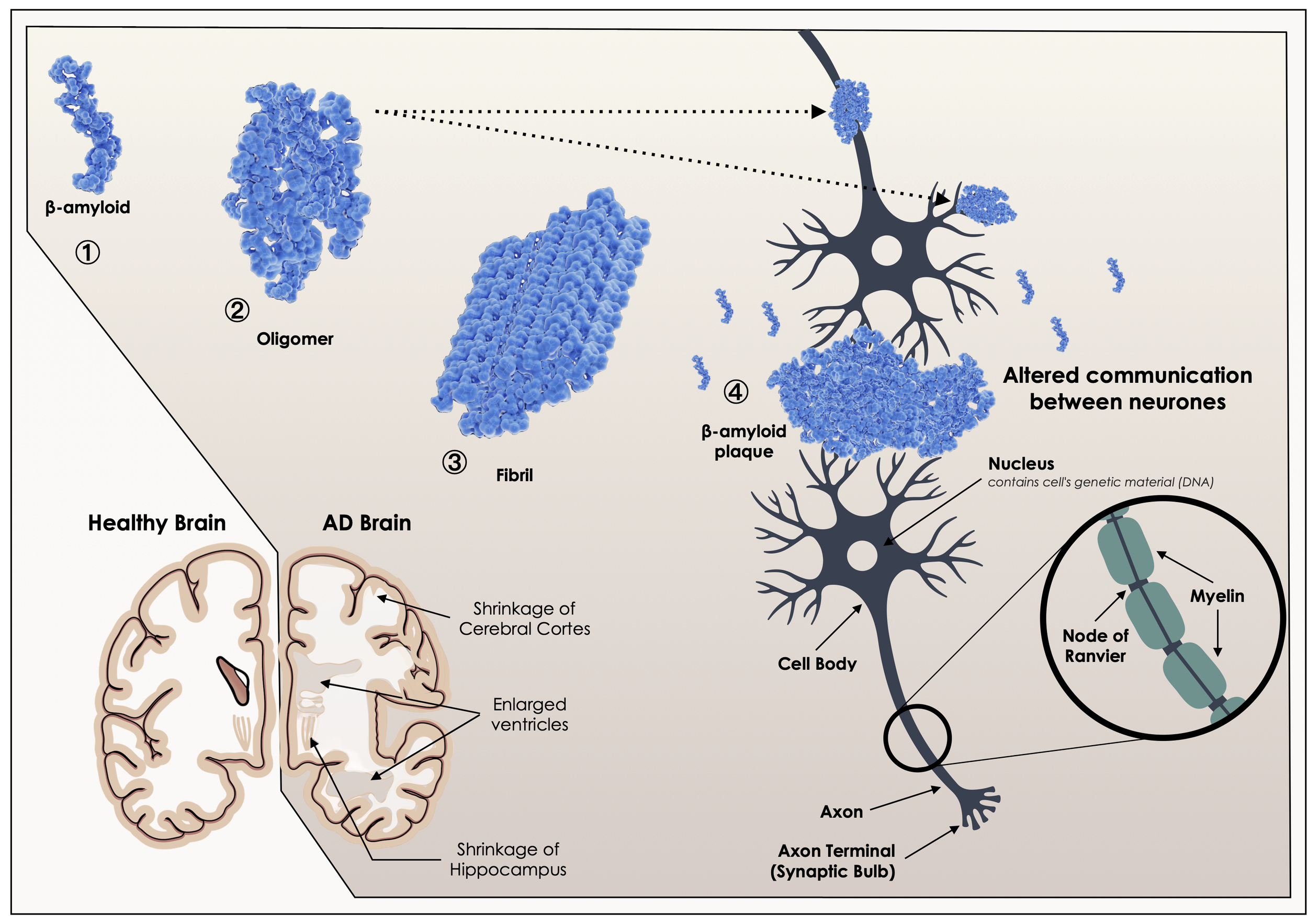
Soluble amyloid-β self-assemble to form soluble toxic aggregates (oligomers), and finally into insoluble fibrils, which subsequently cause synaptic dysfunction (block the transmission of impulse from one neuron to the next) and neuronal death, a hallmark of Alzheimer’s disease (AD)
Neurodegenerative diseases like Huntington’s, Alzheimer’s, and Parkinson’s are all associated with amyloid deposits in the brain. Understanding the toxic impact of oligomers opens new avenues to better understand and find new forms for treatment.
“One of the big mysteries of Huntington’s, Alzheimer’s, and ALS is why disease coincides with amyloid, yet the amyloids themselves are not the main culprits.”
Huntington’s and eight other diseases, collectively called “PolyQ diseases,” occur when certain proteins have a repeat that is too long. Because of the length of the molecule, the protein somehow folds into a specific structure that starts a chain reaction (becoming insoluble fibrils then plaque) that eventually kill brain cells.
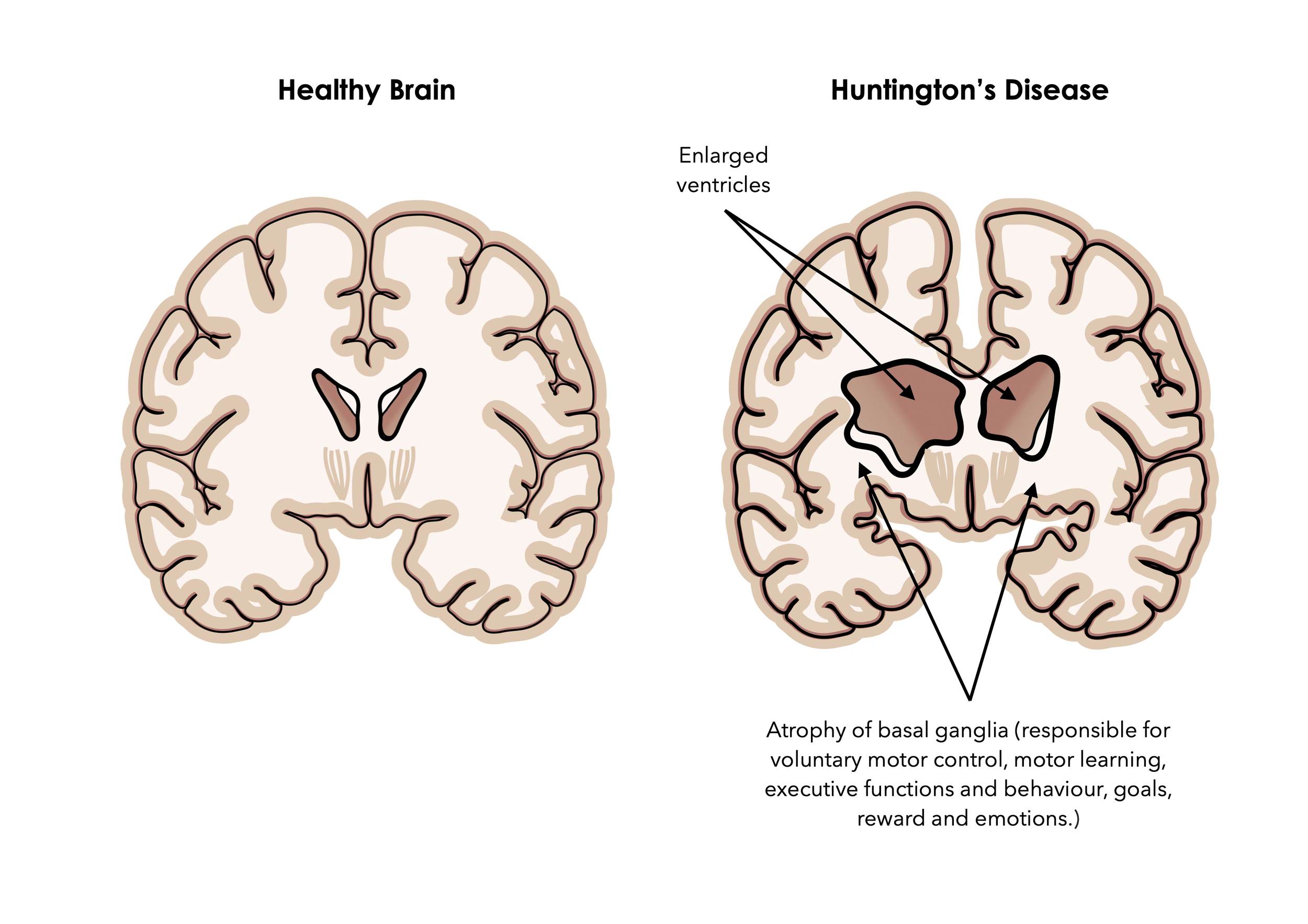
Huntington’s disease is characterised by enlargement of ventricles and atrophy of cerebral nerve tissue and basal ganglia, eventually leading to involuntary movement and muscle twitching, rigidity, speech difficulties, trouble focusing and cognitive impairment, mood swings, behavioural changes, and depression,.
“For three decades, we’ve known that Huntington’s and related fatal diseases occur when proteins contain more than around 36 Qs in a row, causing them to form chains of proteins in the brain, but we didn’t know why.
“We’ve now figured out what the first link in the chain looks like, and, in doing so, have discovered a new way to stop it.”
This is indeed great news because by understanding the cause, rather than creating drugs to address the symptoms with questionable effects, the biological markers can be managed, even rectified, with the potential to slow down the progression or even treat the disease.
“I am, frankly, astonished that such an intuitive physical model of nucleation emerged despite the intrinsic complexity of the cellular environment.
I am truly excited by the intuition and the testable hypotheses that this work inspires.”
While the recently published research only offers some reassurance that new forms of treatment could be developed, it is still a huge step forward, bringing hope to many patients.
“Current therapeutic efforts focused on lowering the levels of mutant Huntingtin have not been successful. We suggest that therapies designed to oligomerize huntingtin preemptively will decelerate the disease.”
The role of Naturopathic Nutrition
Clinical experience and many cases with similar traits have led us to identify several factors that may accelerate the progression of the disease, occur concomitantly, and even exacerbate symptoms.
In 95% of cases, we uncovered dysbiosis and gut inflammation, often due to poor eating habits, restrictive eating (malnutrition, entire food or food groups avoidance), hypochlorhydria (low stomach acid), reflux (nearly all clients were on PPIs or over-the-counter antacids), dyspepsia, brain fog (memory problems, lack of judgement and excitement — nothing matters, including no pleasure in eating), and issues with movement.
In many cases, blood sugar imbalances were also a major issue, and a majority were also positive for SIBO and candidiasis (stool test), dealing with severe bouts of diarrhoea (making it difficult for them to even venture outside, unless they knew where the closest toilets were).
Every client reported suffering from chronic stress, anxiety and/or depression, sleep problems, and feeling constantly tired. They also reported deep forms of distress due to the lack of social interaction or doing the things they loved to do, because their condition had taken over every part of their lives.
Protocol highlights
In our clinic, we offer individualised protocols based on your circumstances, the severity of symptoms and your ability to change your diet and lifestyle and implement new stress-relieving techniques in your day. Therefore, your report will be different from anyone else we have seen in clinics. However, some of the main points that may help you include:
Increase fibre in your diet, particularly soluble (prebiotic) fibre. 3/4 of your plate must be vegetables (preferably, wholesome and minimally processed). Freely add flax/linseeds, chia and hemp seeds to your diet.
Follow an anti-inflammatory diet, made of plenty of fruits and vegetables of as many different colours as possible (5 cups a day minimum of a rainbow diet — pick one vegetable of each colour, which you can eat raw (bitter leaves, fennel, artichokes), minimally cooked (all green vegetables and cabbages) or baked (root vegetables), or juice)
Aim to consume 1-3 portions of small fatty fish (must be wild — avoid farm fish). Think S.M.A.S.H.: sockeye salmon, mackerel, anchovies, sardines and herrings. River trout is also acceptable. If you do not eat fish, supplement with pure fish oil and astaxanthin for a more potent combination. Remember that your brain is made up of fat and that omega-3 fatty acids are shown to have anti-inflammatory properties, which is essential in a brain support protocol.
Exercise daily, at your own pace, as long as it raises your blood pressure and makes you sweat. If you haven’t exercised for a long time, start walking short distances and increasing your regime until you’re comfortable running or doing more physical exercise. Also, consider pilates and yoga for core strengthening.
Some people may benefit from a ketogenic-style diet and consume more healthy ft to support brain function, balance blood glucose and lipid levels, increase insulin sensitivity, and manage weight (must be supervised).
When SIBO or candidiasis are uncovered, you may have to limit your intake of sugar and quick-release carbs, and in some extreme cases and unresolved symptoms, following an Elemental Diet may be necessary. You may also have to address vaginal pH (thrush) and chronic cystitis as part of the protocol.
Aim to consume your (full) calorie allowance to match expenditure. Avoid restrictive eating. Do not remove food groups from your diet without the expert advice of your health practitioner.
Stay hydrated. If you rarely feel thirsty, use apps or reminders to help you drink more.
Implement stress-relieving exercises in your day and use the power of affirmation daily. Use what works best for you. Journalling is also a great tool to use first thing in the morning and last before bed. It can also help you switch off from using electronic devices, checking emails at a moment when your brain should slow down or using social media.
During your consultation, we will address every part of your life, medical history, family history and may recommend certain test to evaluate all contributing factors and the cause(s) of symptoms.
References
1. Kandola, T. et al. (2023). Pathologic polyglutamine aggregation begins with a self-poisoning polymer crystal. eLife. 12: RP86939. doi:10.7554/eLife.86939.1
2. Salahuddin, P. et al. (2016). Structure of amyloid oligomers and their mechanisms of toxicities: Targeting amyloid oligomers using novel therapeutic approaches. European Journal of Medicinal Chemistry, 114, pp. 41-58. doi:10.1016/j.ejmech.2016.02.065
3. Cohen-Carmon, D. Meshorer, E. (2012). Polyglutamine (polyQ) disorders: The chromatin connection. Nucleus. 3(5), pp. 433-441. doi:10.4161/nucl.21481.
4. Glenner, GG. Wong, CW. (1984). Alzheimer's disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications. 120(3), pp. 885-890. doi:10.1016/s0006-291x(84)80190-4
5. Shao, F. et al. (2022). Microglia and neuroinflammation: Crucial pathological mechanisms in traumatic brain injury-induced neurodegeneration. Frontiers in Aging Neuroscience. 14:825086. doi:10.3389/fnagi.2022.825086
6. Edwards, JD. Ramirez, J. Black, SE. (2015). Unravelling the potential co-contributions of cerebral small vessel vasculopathy to the pathogenesis of Alzheimer's dementia. Alzheimer's Research & Therapy. 7(1), 49. doi:10.1186/s13195-015-0133-2
7. Badimon, A. et al. (2020). Negative feedback control of neuronal activity by microglia. Nature. 586, pp. 417–423. doi:10.1038/s41586-020-2777-8
8. Cserép, C. et al. (2020). Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 367(6477), pp. 528-537. doi:10.1126/science.aax6752
9. Borst, K. Dumas, AA. Prinz, M. (2021). Microglia: Immune and non-immune functions. Immunity. 54(10), pp. 2194-2208. doi:10.1016/j.immuni.2021.09.014
10. Vainchtein, ID. et al. (2023). Characterizing microglial gene expression in a model of secondary progressive multiple sclerosis. Glia. 71(3), pp. 588-601. doi:10.1002/glia.24297
11. Gomez, W. et al. (2020). Down syndrome and Alzheimer's disease: common molecular traits beyond the amyloid precursor protein. Aging. 12(1), pp. 1011-1033. doi.org/10.18632/aging.102677